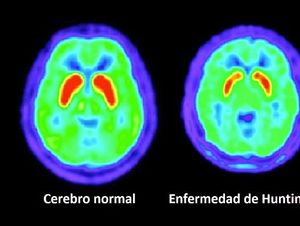
Es un trastorno en el cual las neuronas en ciertas partes del cerebro se desgastan o se degeneran. La enfermedad se transmite de padres a hijos.
Causas
La enfermedad de Huntington es causada por un defecto genético en el cromosoma N.° 4. El defecto hace que una parte del ADN ocurra muchas más veces de las debidas.
El defecto se llama repetición CAG. Normalmente, esta sección del ADN se repite de 10 a 28 veces, pero en una persona con la enfermedad de Huntington, se repite de 36 a 120 veces.
A medida que el gen se transmite de padres a hijos, el número de repeticiones tiende a ser más grande. Cuanto mayor sea el número de repeticiones, mayor será la posibilidad de que una persona presente síntomas a una edad más temprana. Por lo tanto, como la enfermedad se transmite de padres a hijos, los síntomas se desarrollan a edades cada vez más tempranas.
Hay dos formas de la enfermedad de Huntington:
La más común es la de aparición en la edad adulta. Las personas con esta forma de la enfermedad generalmente presentan síntomas a mediados de la tercera y cuarta década de sus vidas.
Una forma de la enfermedad de Huntington de aparición temprana representa un pequeño número de personas y se inicia en la niñez o en la adolescencia.
Si uno de sus padres tiene la enfermedad de Huntington, usted tiene un 50% de probabilidad de recibir el gen.
Si usted recibe el gen de sus padres, también puede transmitir el gen a sus hijos, quienes también tendrán un 50% de probabilidades de heredar el gen. Si usted no recibe el gen de sus padres, no es posible para que usted pueda pasar el gen a sus hijos.
Síntomas
Los cambios de comportamiento pueden ocurrir antes de los problemas de movimiento y pueden incluir:
Comportamientos antisociales
Alucinaciones
Irritabilidad
Malhumor
Inquietud o impaciencia
Paranoia
Psicosis
Los movimientos anormales e inusuales incluyen:
Movimientos faciales, incluso muecas
Girar la cabeza para cambiar la posición de los ojos
Movimientos espasmódicos rápidos y súbitos de los brazos, las piernas, la cara y otras partes del cuerpo
Movimientos lentos e incontrolables
Marcha inestable
Demencia que empeora lentamente, incluso:
Desorientación o confusión
Pérdida de la capacidad de discernimiento
Pérdida de la memoria
Cambios de personalidad
Cambios en el lenguaje
Los síntomas adicionales que pueden estar asociados con esta enfermedad son:
Ansiedad, estrés y tensión
Dificultad para deglutir
Deterioro del habla
Síntomas en los niños:
Rigidez
Movimientos lentos
Temblor
Pruebas y exámenes
El médico llevará a cabo un examen físico y puede hacer preguntas acerca de los síntomas y los antecedentes familiares del paciente. También se llevará a cabo un examen del sistema nervioso. El médico puede ver signos de:
Demencia
Movimientos anormales
Reflejos anormales
Marcha amplia y con "pavoneo"
Lenguaje dubitativo o articulación deficiente
Otros exámenes que pueden mostrar signos de enfermedad de Huntington son:
Pruebas psicológicas
Resonancia magnética o tomografía computarizada de la cabeza
TEP (isótopos) del cerebro
Hay disponibilidad de pruebas genéticas para determinar si una persona es portadora del gen de la enfermedad de Huntington.
Tratamiento
No existe cura para la enfermedad de Huntington y no hay una forma conocida de detener el empeoramiento de la enfermedad. El objetivo del tratamiento es reducir los síntomas y ayudar a la persona a valerse por sí misma por el mayor tiempo posible.
Se pueden recetar medicamentos según los síntomas.
Los bloqueadores de la dopamina pueden ayudar a reducir los comportamientos y movimientos anormales.
Medicamentos como tetrabenazina y amantidina se usan para tratar de controlar los movimientos adicionales.
La depresión y el suicidio son comunes entre las personas con enfermedad de Huntington. Es importante que los cuidadores vigilen los síntomas y busquen ayuda médica para la persona de inmediato.
A medida que la enfermedad progrese, la persona necesitará asistencia y supervisión. Con el paso del tiempo, es posible que requiera atención durante las 24 horas.
Grupos de apoyo
Huntington's Disease Society of America -- hdsa.org
Expectativas (pronóstico)
La enfermedad de Huntington causa discapacidad que empeora con el tiempo. Las personas que padecen esta enfermedad generalmente mueren al cabo de 15 a 20 años. Con frecuencia, la causa de la muerte es una infección. El suicidio también es común.
Es importante tener en cuenta que la enfermedad afecta a todas las personas de manera diferente. El número de copias o repeticiones CAG del gen puede determinar la gravedad de los síntomas. Las personas con pocas copias o repeticiones pueden tener movimientos anormales leves más tarde en la vida y progresión lenta de la enfermedad. Aquellas con un número mayor de repeticiones pueden resultar gravemente afectadas a una edad temprana.
Cuándo contactar a un profesional médico
Consulte con el médico si usted o un miembro de la familia presenta síntomas de este trastorno.
Prevención
Se aconseja la asesoría genética si existen antecedentes familiares de la enfermedad de Huntington. Los expertos también recomiendan la asesoría genética para parejas con antecedentes familiares de esta enfermedad que estén contemplando la posibilidad de tener hijos.
Nombres alternativos
Corea de Huntington
Referencias
Jankovic J. Movement disorders. In: Daroff RB, Fenichel GM, Jankovic J, Mazziotta JC, eds. Bradley's Neurology in Clinical Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2012:chap 71.
Mink JW. Movement disorders. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 597.
Ultima revisión 6/1/2015
Versión en inglés revisada por: Daniel Kantor, MD, Kantor Neurology, Coconut Creek, FL and Immediate Past President of the Florida Society of Neurology (FSN). Review provided by VeriMed Healthcare Network. Also reviewed by David Zieve, MD, MHA, Isla Ogilvie, PhD, and the A.D.A.M. Editorial team.